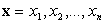
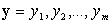
ΒΙΟΠΛΗΡΟΦΟΡΙΚΗ
(«Η/Υ ΣΤΗ ΒΙΟΛΟΓΙΑ»)
ΕΡΓΑΣΤΗΡΙΑΚΗ ΑΣΚΗΣΗ
BLAST-FASTA-CLUSTAL
Εργαλεία αναζήτησης ομοιοτήτων και πολλαπλής στοίχισης ακολουθιών
Ζωή Λίτου, Παντελής Μπάγκος & Σταύρος Ι. Χαμόδρακας
Τομέας Βιολογίας Κυττάρου & Βιοφυσικής
Τμήμα Βιολογίας
Παν/μιο Αθηνών
Φεβρουάριος 2002
Στόχος της άσκησης είναι η εισαγωγή στην χρήση βιολογικών βάσεων ακολουθιών πρωτεϊνών και DNA και προγραμμάτων στοίχισης ακολουθιών χρησιμοποιώντας Web-based μέσα. Η στοίχιση δυο ακολουθιών (pairwise alignment), η πολλαπλή στοίχιση (multiple alignment) καθώς και η αναζήτηση ομόλογων πρωτεϊνών μέσα σε μια μεγάλη βάση δεδομένων, είναι θέματα που απασχολούν χρόνια τους επιστήμονες, και αποκτούν όσο περνάει ο καιρός, όλο και μεγαλύτερη σημασία, καθώς αυξάνεται ραγδαία το μέγεθος των διαθέσιμων βάσεων δεδομένων, αλλά και η ευκολία πρόσβασης σ΄αυτές για τους ερευνητές.
Θα παρουσιαστούν παρακάτω τα βασικά στοιχεία της θεωρίας για στοιχίσεις μακρομορίων (DNA και πρωτεϊνών), και οι βασικοί αλγόριθμοι που χρησιμοποιούνται τόσο για μια προς μια στοίχιση των ακολουθιών (Smith & Waterman, BLAST, FASTA) όσο και για πολλαπλή στοίχιση (CLUSTALW, CLUSTALX). Όπως είναι εμφανές, οι μέθοδοι στοίχισης είναι πολύ χρήσιμοι στον ερευνητή, για να ταυτοποιήσει μια άγνωστη πρωτεΐνη, αλλά και για να εξαγάγει πληροφορίες τόσο εξελικτικές όσο και λειτουργικές.
Το BLAST και το FASTA αποτελούν τους πιο συχνά χρησιμοποιούμενους ευριστικούς (heuristic) αλγορίθμους και βρίσκονται διαθέσιμοι στο διαδίκτυο και σε τοπικές εκδόσεις. Ο όρος ευριστικός (heuristic) χρησιμοποιείται για αλγόριθμους που βρίσκουν λύσεις μεταξύ όλων των πιθανών, αλλά δεν εγγυώνται ότι θα βρουν τη βέλτιστη. Τέτοιου είδους αλγόριθμοι, βρίσκουν μια λύση κοντινή στη βέλτιστη (close to the best one) γρήγορα και εύκολα.
Αν υποθέσουμε ότι έχουμε 2 ακολουθίες πρωτεϊνών μήκους η.
αποδεικνύεται ότι οι πιθανές στοιχίσεις των δυο αυτών ακολουθιών είναι:
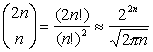 (1)
(1)
Ο αριθμός αυτός είναι τεράστιος ακόμα και για μικρές ακολουθίες, και γι' αυτό το λόγο έχουν αναπτυχθεί αλγόριθμοι δυναμικού προγραμματισμού οι οποίοι βρίσκουν την καλύτερη στοίχιση των ακολουθιών στο μικρότερο δυνατό χρόνο (Smith & Waterman, 1981). Για να γίνει η στοίχιση αυτή χρησιμοποιείται ένα είδος score, το οποίο μετράει την ομοιότητα τον αμινοξέων. Στην πιο απλή περίπτωση χρησιμοποιούμε το παρακάτω score
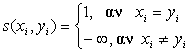 (2)
(2)
Αντίστοιχες συνεισφορές στο score (αρνητικές) έχουν οι προσθήκες κενών, ενώ αξίζει να αναφερθεί ότι άλλη ποινή ορίζουμε για την προσθήκη κενών (gap open penalty) και άλλη για την επέκταση τους (gap extension penalty). Συνηθίζεται πλέον, εκτός αν υπάρχουν ειδικοί λόγοι, να μην χρησιμοποιείται ο παραπάνω τρόπος υπολογισμού του score, αλλά να λαμβάνουμε υπόψη μας και τις εξελικτικές αλλά και λειτουργικές σχέσεις των αμινοξέων, και αυτό πραγματοποιείται με τη χρήση των πινάκων υποκαταστάσεως.
Μια από τις πιο σημαντικές παραμέτρους που πρέπει να λαμβάνουμε υπ' όψη μας στη χρήση των προγραμμάτων τοπικής στοίχισης, είναι το είδος του πίνακα υποκατάστασης (substitution matrix) που χρησιμοποιούμε και το ύψος της ποινής για τα κενά που ενδέχεται να εισαχθούν (gap penalties), για να γίνει η στοίχιση.
Ανεξαρτήτως του αλγορίθμου που χρησιμοποιούμε, σε γενικές γραμμές, το μέγεθος το οποίο καθορίζει τη στοίχιση είναι το score, το οποίο παίρνει κάποια τιμή θετική ή αρνητική για κάθε αντιστοίχιση των αμινοξέων, από κάποιο 20Χ20 πίνακα υποκαταστάσεως. Οι πίνακες αυτοί [PAM (Point Accepted Mutation), BLOSUM (BLOcks SUbstitution Matrix) κλπ] εκφράζουν σε γενικές γραμμές την εξελικτική σχέση των 20 αμινοξέων, π.χ. η βαλίνη και η ισολευκίνη θα έχουν σχεδόν πάντα θετική συνεισφορά στο score, αν συναντηθούν σαν ζεύγος, ενώ η αργινίνη και η φενυλαλανίνη αρνητική. Πρέπει να τονιστεί ότι ο κάθε πίνακας είναι φτιαγμένος για να πραγματοποιεί καλύτερες στοιχίσεις, σε μια δεδομένη εξελικτική σχέση των πρωτεϊνών. 'Aλλος δηλαδή πίνακας ταιριάζει στην περίπτωση που έχουμε δυο πολύ ομόλογες πρωτεΐνες και άλλος για δυο πρωτεΐνες πολύ απομακρυσμένες εξελικτικά. Σε μια αναζήτηση όμως έναντι ολόκληρης της βάσης είναι καλύτερο να χρησιμοποιούμε τους πιο αποδεκτούς πίνακες, όπως τον BLOSUM62.
Όσον αφορά στην ποινή για τα κενά, συνηθίζεται να ορίζουμε μεγαλύτερη ποινή για το πρώτο κενό (gap open penalty) και μικρότερη για τα επόμενα (gap extension penalty). Είναι προφανές ότι μικρές ή μηδενικές ποινές θα οδηγούν σε ολικές στοιχίσεις ενώ πολύ μεγάλες αρνητικές
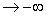 θα οδηγούν σε πιο «αυστηρές» στοιχίσεις, χωρίς κενά. Παρακάτω δίνονται οι δυο πιο γνωστοί πίνακες υποκαταστάσεως ο BLOSUM62, και ο PAM250, όπου βλέπουμε τα παρόμοια (εξελικτικά και φυσικοχημκά) αμινοξέα να δίνουν θετικό score αν συναντηθούν ως ζεύγος ενώ τα διαφορετικά να δίνουν αρνητικό. Παρατηρείστε ότι παρ' όλο που δίνουν ανάλογα αποτελέσματα αυτά δεν είναι εντελώς όμοια. Η διαφορά των δυο πινάκων φαίνεται απο τα δύο μεγέθη Entropy (σχετική εντροπια - Kullback & Leibler) και απο το Expected (αναμενόμενο score ανά κατάλοιπο).
θα οδηγούν σε πιο «αυστηρές» στοιχίσεις, χωρίς κενά. Παρακάτω δίνονται οι δυο πιο γνωστοί πίνακες υποκαταστάσεως ο BLOSUM62, και ο PAM250, όπου βλέπουμε τα παρόμοια (εξελικτικά και φυσικοχημκά) αμινοξέα να δίνουν θετικό score αν συναντηθούν ως ζεύγος ενώ τα διαφορετικά να δίνουν αρνητικό. Παρατηρείστε ότι παρ' όλο που δίνουν ανάλογα αποτελέσματα αυτά δεν είναι εντελώς όμοια. Η διαφορά των δυο πινάκων φαίνεται απο τα δύο μεγέθη Entropy (σχετική εντροπια - Kullback & Leibler) και απο το Expected (αναμενόμενο score ανά κατάλοιπο).
# BLOSUM Clustered Scoring Matrix in 1/2 Bit Units # Cluster Percentage: >= 62 # Entropy = 0.6979, Expected = -0.5209 A R N D C Q E G H I L K M F P S T W Y V B Z X * A 4 -1 -2 -2 0 -1 -1 0 -2 -1 -1 -1 -1 -2 -1 1 0 -3 -2 0 -2 -1 0 -4 R -1 5 0 -2 -3 1 0 -2 0 -3 -2 2 -1 -3 -2 -1 -1 -3 -2 -3 -1 0 -1 -4 N -2 0 6 1 -3 0 0 0 1 -3 -3 0 -2 -3 -2 1 0 -4 -2 -3 3 0 -1 -4 D -2 -2 1 6 -3 0 2 -1 -1 -3 -4 -1 -3 -3 -1 0 -1 -4 -3 -3 4 1 -1 -4 C 0 -3 -3 -3 9 -3 -4 -3 -3 -1 -1 -3 -1 -2 -3 -1 -1 -2 -2 -1 -3 -3 -2 -4 Q -1 1 0 0 -3 5 2 -2 0 -3 -2 1 0 -3 -1 0 -1 -2 -1 -2 0 3 -1 -4 E -1 0 0 2 -4 2 5 -2 0 -3 -3 1 -2 -3 -1 0 -1 -3 -2 -2 1 4 -1 -4 G 0 -2 0 -1 -3 -2 -2 6 -2 -4 -4 -2 -3 -3 -2 0 -2 -2 -3 -3 -1 -2 -1 -4 H -2 0 1 -1 -3 0 0 -2 8 -3 -3 -1 -2 -1 -2 -1 -2 -2 2 -3 0 0 -1 -4 I -1 -3 -3 -3 -1 -3 -3 -4 -3 4 2 -3 1 0 -3 -2 -1 -3 -1 3 -3 -3 -1 -4 L -1 -2 -3 -4 -1 -2 -3 -4 -3 2 4 -2 2 0 -3 -2 -1 -2 -1 1 -4 -3 -1 -4 K -1 2 0 -1 -3 1 1 -2 -1 -3 -2 5 -1 -3 -1 0 -1 -3 -2 -2 0 1 -1 -4 M -1 -1 -2 -3 -1 0 -2 -3 -2 1 2 -1 5 0 -2 -1 -1 -1 -1 1 -3 -1 -1 -4 F -2 -3 -3 -3 -2 -3 -3 -3 -1 0 0 -3 0 6 -4 -2 -2 1 3 -1 -3 -3 -1 -4 P -1 -2 -2 -1 -3 -1 -1 -2 -2 -3 -3 -1 -2 -4 7 -1 -1 -4 -3 -2 -2 -1 -2 -4 S 1 -1 1 0 -1 0 0 0 -1 -2 -2 0 -1 -2 -1 4 1 -3 -2 -2 0 0 0 -4 T 0 -1 0 -1 -1 -1 -1 -2 -2 -1 -1 -1 -1 -2 -1 1 5 -2 -2 0 -1 -1 0 -4 W -3 -3 -4 -4 -2 -2 -3 -2 -2 -3 -2 -3 -1 1 -4 -3 -2 11 2 -3 -4 -3 -2 -4 Y -2 -2 -2 -3 -2 -1 -2 -3 2 -1 -1 -2 -1 3 -3 -2 -2 2 7 -1 -3 -2 -1 -4 V 0 -3 -3 -3 -1 -2 -2 -3 -3 3 1 -2 1 -1 -2 -2 0 -3 -1 4 -3 -2 -1 -4 B -2 -1 3 4 -3 0 1 -1 0 -3 -4 0 -3 -3 -2 0 -1 -4 -3 -3 4 1 -1 -4 Z -1 0 0 1 -3 3 4 -2 0 -3 -3 1 -1 -3 -1 0 -1 -3 -2 -2 1 4 -1 -4 X 0 -1 -1 -1 -2 -1 -1 -1 -1 -1 -1 -1 -1 -1 -2 0 0 -2 -1 -1 -1 -1 -1 -4 * -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 1 |
# This matrix was produced by "pam" Version 1.0.6 [28-Jul-93] # PAM 250 substitution matrix, scale = ln(2)/3 = 0.231049 # Expected score = -0.844, Entropy = 0.354 bits # Lowest score = -8, Highest score = 17 A R N D C Q E G H I L K M F P S T W Y V B Z X * A 2 -2 0 0 -2 0 0 1 -1 -1 -2 -1 -1 -3 1 1 1 -6 -3 0 0 0 0 -8 R -2 6 0 -1 -4 1 -1 -3 2 -2 -3 3 0 -4 0 0 -1 2 -4 -2 -1 0 -1 -8 N 0 0 2 2 -4 1 1 0 2 -2 -3 1 -2 -3 0 1 0 -4 -2 -2 2 1 0 -8 D 0 -1 2 4 -5 2 3 1 1 -2 -4 0 -3 -6 -1 0 0 -7 -4 -2 3 3 -1 -8 C -2 -4 -4 -5 12 -5 -5 -3 -3 -2 -6 -5 -5 -4 -3 0 -2 -8 0 -2 -4 -5 -3 -8 Q 0 1 1 2 -5 4 2 -1 3 -2 -2 1 -1 -5 0 -1 -1 -5 -4 -2 1 3 -1 -8 E 0 -1 1 3 -5 2 4 0 1 -2 -3 0 -2 -5 -1 0 0 -7 -4 -2 3 3 -1 -8 G 1 -3 0 1 -3 -1 0 5 -2 -3 -4 -2 -3 -5 0 1 0 -7 -5 -1 0 0 -1 -8 H -1 2 2 1 -3 3 1 -2 6 -2 -2 0 -2 -2 0 -1 -1 -3 0 -2 1 2 -1 -8 I -1 -2 -2 -2 -2 -2 -2 -3 -2 5 2 -2 2 1 -2 -1 0 -5 -1 4 -2 -2 -1 -8 L -2 -3 -3 -4 -6 -2 -3 -4 -2 2 6 -3 4 2 -3 -3 -2 -2 -1 2 -3 -3 -1 -8 K -1 3 1 0 -5 1 0 -2 0 -2 -3 5 0 -5 -1 0 0 -3 -4 -2 1 0 -1 -8 M -1 0 -2 -3 -5 -1 -2 -3 -2 2 4 0 6 0 -2 -2 -1 -4 -2 2 -2 -2 -1 -8 F -3 -4 -3 -6 -4 -5 -5 -5 -2 1 2 -5 0 9 -5 -3 -3 0 7 -1 -4 -5 -2 -8 P 1 0 0 -1 -3 0 -1 0 0 -2 -3 -1 -2 -5 6 1 0 -6 -5 -1 -1 0 -1 -8 S 1 0 1 0 0 -1 0 1 -1 -1 -3 0 -2 -3 1 2 1 -2 -3 -1 0 0 0 -8 T 1 -1 0 0 -2 -1 0 0 -1 0 -2 0 -1 -3 0 1 3 -5 -3 0 0 -1 0 -8 W -6 2 -4 -7 -8 -5 -7 -7 -3 -5 -2 -3 -4 0 -6 -2 -5 17 0 -6 -5 -6 -4 -8 Y -3 -4 -2 -4 0 -4 -4 -5 0 -1 -1 -4 -2 7 -5 -3 -3 0 10 -2 -3 -4 -2 -8 V 0 -2 -2 -2 -2 -2 -2 -1 -2 4 2 -2 2 -1 -1 -1 0 -6 -2 4 -2 -2 -1 -8 B 0 -1 2 3 -4 1 3 0 1 -2 -3 1 -2 -4 -1 0 0 -5 -3 -2 3 2 -1 -8 Z 0 0 1 3 -5 3 3 0 2 -2 -3 0 -2 -5 0 0 -1 -6 -4 -2 2 3 -1 -8 X 0 -1 0 -1 -3 -1 -1 -1 -1 -1 -1 -1 -1 -2 -1 0 0 -4 -2 -1 -1 -1 -1 -8 * -8 -8 -8 -8 -8 -8 -8 -8 -8 -8 -8 -8 -8 -8 -8 -8 -8 -8 -8 -8 -8 -8 -8 1 |
'Eνα πολύ μεγάλο θέμα που προκύπτει στη στοίχιση ακολουθιών είναι η εύρεση της Στατιστικής Σημαντικότητας, των αποτελεσμάτων. Συγκεκριμμένα μας ενδιαφέρει το πως μπορούμε να διαχωρίσουμε «τυχαία» ευρήματα από «σημαντικά». Το P-value ενός στατιστικού ελέγχου (γιατι περί τέτοιου πρόκειται) είναι η πιθανότητα, ένα αποτέλεσμα τόσο ακραίο ή και περισσότερο, να έχει προκύψει κατα τύχη δεδομένου ότι ισχύει η μηδενική υπόθεση (στην περίπτωση μας, ότι οι δύο ακολουθίες που συγκρίναμε δεν έχουν καμία σχέση μεταξύ τους). Είναι προφανές ότι αναφερόμαστε σε παραμετρικό έλεγχο και χρειάζεται να ξέρουμε την κατανομή που ακολουθεί η τυχαία μεταβλητή που μας ενδιαφέρει, το score δηλαδή.
'Eχει αποδειχθεί (Altschul and Gish,1996) ότι η τυχαία μεταβλητή του score όταν αναφερόμαστε σε τοπικές στοιχίσεις, ακολουθεί ασυμπτωτικά την κατανομή των ακραίων τιμών (Extreme Value Distribution-EVD) η οποία έχει α.σ.κ. (αθροιστική συνάρτηση κατανομής) :
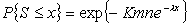 (3)
(3)
όπου m,n τα μήκη των υπό σύγκριση ακολουθιών και Κ, λ οι παράμετροι της κατανομής. Στην περίπτωση που συγκρίνουμε μια ακολουθία μήκους n, με μια ολόκληρη βάση δεδομένων, τότε το m είναι το συνολικό άθροισμα των αμινοξέων, που περιέχονται στις πρωτεϊνες της βάσης. Η πιθανότητα (P-value) να βρεθεί μια τιμή τοσο ακραία, η και πιο ακραία απο x θα είναι
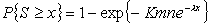 (4)
(4)
Η ίδια η ύπαρξη των HSP (High-scoring Segment Pair), σαν σπάνια ενδεχόμενα περιγράφεται απο την κατανομή Poisson με αναμενόμενη τιμή (E-value) να δίνεται από τη σχέση :
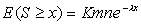 (5)
(5)
Η τιμή αυτή συμβολίζει την αναμενόμενη (expected) τιμή για την εμφάνιση περιοχών με score τουλάχιστο ίσο με x στη βάση δεδομένων που μελετάμε. Επειδή η τιμή του score εξαρτάται αποκλειστικά απο τον πίνακα υποκατάστασης (substitution matrix) π.χ. PAM, BLOSUM κλπ, και από τις ποινές για την εμφάνιση των κενών (gap penalties) δεν μας προσφέρει χρήσιμα συμπεράσματα, καθώς δύο αποτελέσματα που έχουν προκύψει από διαφορετικό σύνολο παραμέτρων δεν είναι συγκρίσιμα. Έτσι είναι προτιμότερο να χρησιμοποιούμε μια «κανονικοποιημένη» κλίμακα ως εξής:
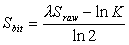 (6)
(6)
όπου
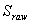 είναι το score που υπολογίστηκε με συγκεκριμένες παραμέτρους, και Κ, λ οι παράμετροι της κατανομής που είδαμε παραπάνω. Έτσι η αναμενόμενη τιμή για την εμφάνιση των HSP, είναι πλέον :
είναι το score που υπολογίστηκε με συγκεκριμένες παραμέτρους, και Κ, λ οι παράμετροι της κατανομής που είδαμε παραπάνω. Έτσι η αναμενόμενη τιμή για την εμφάνιση των HSP, είναι πλέον :
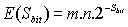 (7)
(7)
Πρέπει να τονιστεί εδώ ότι καθώς ισχύει η προσέγγιση :
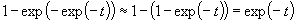 (8)
(8)
και καθώς
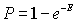 (σχ. 3,4,5), αν το E-value είναι πολύ μικρό, τότε
(σχ. 3,4,5), αν το E-value είναι πολύ μικρό, τότε
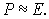
'Eτσι το BLAST χρησιμοποιεί μόνο το E-value, το οποίο γίνεται πιο εύκολα κατανοητό, απο τους βιολόγους, και επι πλέον πέρνει τιμές απο 0 εώς τον αριθμό των ακολουθιών στη βάση. Όσον αφορά την εκτίμηση των παραμέτρων Κ και λ, το BLAST χρησιμοποιεί εκτιμητές μέγιστης πιθανοφάνειας, από μια προ-κατασκευασμένη βάση αντίστοιχης σύστασης, και για το λόγο αυτό είναι και πιο γρήγορο απο το FASTA, το οποίο χρησιμοποιεί τα αποτελέσματα από τις ίδιες τις πρωτεΐνες της βάσης στην οποία κάνει τον έλεγχο.
Αξίζει να σημειωθεί ότι στις αρχικές εκδόσεις του BLAST, δεν επιτρεπόταν στοιχιση με κενά, αλλά αυτό τροποποιήθηκε στην βελτιωμένη έκδοση. Όταν αναφερόμαστε σε τέτοιες στοιχίσεις δεν υπάρχουν μαθηματικές αποδείξεις για την κατανομή του score, αλλά όλες οι ενδείξεις συγκλίνουν στο ότι και σ' αυτή την περίπτωση ισχύουν τα παραπάνω αναφερθέντα για τη στατιστική σημαντικότητα (Arratia and Waterman, 1994; Pearson and Wood, 2001).
Κάτι που πρέπει να τονίσουμε τέλος είναι ότι το BLAST, όσον αφορά στο μήκος της βάσης δεδομένων (m) αλλα και της δοθείσας ακολουθίας (n), κάνει έναν ειδικό υπολογισμό και τα ελλατώνει κατά ένα ποσοστό, έτσι ώστε να ανταπεξέλθει στο λεγόμενο "edge effect". Το τελευταίο σημαίνει ότι καθώς μια στοίχιση προχωράει και πλησιάζει στο τέλος τη ακολουθίας, ελλατώνονται οι πιθανοί συνδιασμοί αμινοξέων που μπορούν να προστεθούν, και άρα αλλοιώνονται τα αποτελέσματα για τη στατιστική σημαντικότητα. Έτσι τα τροποποιημένα μεγέθη είναι:
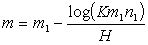 και
και
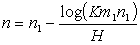 (9)
(9)
όπου m1, n1 τα αρχικά μήκη, Κ η σταθερά της κατανομής EVD, και Η η σχετική εντροπία του πίνακα υποκατάστασης.
Παρακάτω φαίνεται ενα δείγμα από output του BLAST για 2 ακολουθίες, όπου γραμοσκιασμένα φαίνονται τα ονόματα των ακολουθιών, το score και το Ε-value (expect), οι παράμετροι της κατανομής (Κ,λ), ο πίνακας (BLOSUM62) και οι ποινές για τα κενά. Φαίνονται επίσης τα αρχικά m1, n1 (length of database =279182324, length of query =367) και τα τροποποιημένα (m, n) (effective length of database=186375216, effective length of query=245), ενώ εμφανίζεται και το γινόμενο τους m*n (effective search space =45661927920)
Sequence 1 sp|P06996|OMPC_ECOLI Outer membrane protein C precursor Length367(1 .. 367)
Sequence 2 sp|P21420|NMPC_ECOLI Outer membrane porin protein NMPC Length365(1 .. 365)
NOTE:The statistics (bitscore and expect value) is calculated based on the size of nr database
Score = 444 bits (1143), Expect = e-124
Identities = 226/351 (64%), Positives = 260/351 (73%), Gaps = 20/351 (5%)
Query: 25 YNKDGNKLDLYGKVDGLHYFSDNKDVDGDQTYMRLGFKGETQVTDQLTGYGQWEYQIQGN 84
YNKD NKLDLYGKV+ HYFS N DGD TY RLGFKGETQ+ DQLTG+GQWEY+ +GN
Sbjct: 27 YNKDSNKLDLYGKVNAKHYFSSNDADDGDTTYARLGFKGETQINDQLTGFGQWEYEFKGN 86
Query: 85 SAENENNS--WTRVAFAGLKFQDVGSFDYGRNYGVVYDVTSWTDVLPEFGGDTY-GSDNF 141
AE++ +S TR+AFAGLKF D GS DYGRNYGV YD+ +WTDVLPEFGGDT+ +D F
Sbjct: 87 RAESQGSSKDKTRLAFAGLKFGDYGSIDYGRNYGVAYDIGAWTDVLPEFGGDTWTQTDVF 146
Query: 142 MQQRGNGFATYRNTDFFGLVDGLNFAVQYQGKNGNPSGEGFTSGVTNNGRDALRQNGDGV 201
M QR G ATYRN DFFGLVDGLNFA QYQGKN + +T G NGDG
Sbjct: 147 MTQRATGVATYRNNDFFGLVDGLNFAAQYQGKNDRSDFDNYTEG-----------NGDGF 195
Query: 202 GGSITYDYEGFGIGGAISSSKRTDAQNTAAYI-----GNGDRAETYTGGLKYDANNIYLA 256
G S TY+YEGFGIG + S RTD Q A + +G AE + GLKYDANNIYLA
Sbjct: 196 GFSATYEYEGFGIGATYAKSDRTDTQVNAGKVLPEVFASGKNAEVWAAGLKYDANNIYLA 255
Query: 257 AQYTQTYNATRVGSLGWANKAQNFEAVAQYQFDFGLRPSLAYLQSKGKNLGRGYDDEDIL 316
Y++T N T ANKAQNFEAVAQYQFDFGLRPS+AYLQSKGK+LG + D+D++
Sbjct: 256 TTYSETQNMTVFADHFVANKAQNFEAVAQYQFDFGLRPSVAYLQSKGKDLG-VWGDQDLV 314
Query: 317 KYVDVGATYYFNKNMSTYVDYKINLLDDNQFTRDAGINTDNIVALGLVYQF 367
KYVDVGATYYF KNMST+VDYKINLLD N FT+ G++TD+IVA+GLVYQF
Sbjct: 315 KYVDVGATYYFKKNMSTFVDYKINLLDKNDFTKALGVSTDDIVAVGLVYQF 365
Lambda K H
0.315 0.137 0.408
Gapped
Lambda K H
0.267 0.0410 0.140
Matrix: BLOSUM62
Gap Penalties: Existence: 11, Extension: 1
Number of Hits to DB: 2095
Number of Sequences: 0
Number of extensions: 207
Number of successful extensions: 6
Number of sequences better than 10.0: 1
Number of HSP's better than 10.0 without gapping: 1
Number of HSP's successfully gapped in prelim test: 0
Number of HSP's that attempted gapping in prelim test: 0
Number of HSP's gapped (non-prelim): 1
length of query: 367
length of database: 279,182,324
effective HSP length: 122
effective length of query: 245
effective length of database: 186,375,216
effective search space: 45661927920
effective search space used: 45661927920
T: 9
A: 40
X1: 16 ( 7.3 bits)
X2: 129 (49.7 bits)
X3: 129 (49.7 bits)
S1: 42 (22.0 bits)
S2: 72 (32.3 bits)
|
Είναι ο πρώτος ευριστικός (heuristic) αλγόριθμος ευρείας χρήσεως αναζήτησης ομοιοτήτων σε βιολογικές βάσεις. Συγκρίνει μια πρωτεϊνική ακολουθία με μια άλλη πρωτεϊνική ακολουθία ή με μια πρωτεϊνική βάση δεδομένων. Καθώς επίσης και μια DNA ακολουθία με μια άλλη ακολουθία DNA ή με μια DNA βάση.
Το πρόγραμμα FASTA3 βρίσκεται στο site του EBI (European Bioinformatics Institute) ( http://www.ebi.ac.uk/fasta33/ ). Ψάχνει να βρει τις βέλτιστες τοπικές στοιχίσεις από την ανίχνευση της ακολουθίας για μικρές αντιστοιχίες που ονομάζονται "λέξεις" (words). Αρχικά, υπολογίζεται το σύνολο των τμημάτων ("init1") στα οποία υπάρχουν πολλαπλές "λέξεις" (words). Τα αποτελέσματα διάφορων τμημάτων μπορούν να αθροιστούν για να παράγουν ένα "initn". Μια βελτιστοποιημένη στοίχιση που περιλαμβάνει ανοίγματα (gaps) εμφανίζεται ως "opt". Η ευαισθησία και η ταχύτητα της αναζήτησης είναι αντιστρόφως σχετιζόμενες και ελεγχόμενες από τη μεταβλητή "k-tup" που προσδιορίζει το μέγεθος μιας λέξης (word) (Pearson and Lipman, 1988). Το FASTA υπολογίζει τη στατιστική σημαντικότητα "επί τόπου" (on the fly) από το σύνολο των δεδομένων, (πιο ακριβές αλλά έχει προβλήματα αν το σύνολο των δεδομένων είναι πολύ μικρό).
fasta3, - εξερευνεί μια πρωτεϊνική ή νουκλεοτιδική (DNA) βάση ακολουθιών για παρόμοιες ακολουθίες.
fastx/y3 - συγκρίνει μια νουκλεοτιδική ακολουθία (DNA) μεταφρασμένη σε όλα τα πλαίσια ανάγνωσης (reading frames) με μια βάση πρωτεϊνικών ακολουθιών του NCBI.
tfastx/y3 - συγκρίνει μια πρωτεΐνη με μια μεταφρασμένη DNA βάση δεδομένων.
fastf3 - συγκρίνει μείγμα πεπτιδίων με μια πρωτεϊνική βάση δεδομένων.
bic_sw - συγκρίνει μια πρωτεϊνική ή νουκλεοτιδική (DNA) ακολουθία με μια βάση ακολουθιών χρησιμοποιώντας τον αλγόριθμο των Smith-Waterman. (Smith & Waterman, 1981)
Ο αλγόριθμος BLAST χρησιμοποιείται για τη σύγκριση μιας ακολουθίας με μια βάση δεδομένων, έχει αρχικά αναπτυχθεί και διατηρείται από το NCBI (National Center for Biotechnology Information) ( http://www.ncbi.nlm.nih.gov/BLAST/ ) (αλλά λόγω του ότι διατίθεται ελεύθερα υπάρχουν εκδόσεις του και σε άλλες αντίστοιχες βάσεις. Είναι ένας ευριστικός (heuristic) αλγόριθμος σύγκρισης ακολουθιών βελτιστοποιημένης ταχύτητας που χρησιμοποιείται για να ψάχνει σε βάσεις ακολουθιών την άριστη τοπική στοίχιση με μια αναζήτηση. Η αρχική αναζήτηση γίνεται για μια λέξη μήκους "W" ( 3 στο blastp) που δίνει αποτελέσματα για τουλάχιστον "T", όταν συγκρίνεται με την ζητούμενη ακολουθία, χρησιμοποιώντας έναν δεδομένο πίνακα υποκαταστάσεων (substitution matrix). Οι επιτυχείς λέξεις που έχουν score T ή μεγαλύτερο επεκτείνονται και προς τις δύο κατευθύνσεις σε μια απόπειρα να παραχθούν στοιχίσεις που να υπερβαίνουν το προκαθορισμένο κατώφλι (threshold) "S". Οι περιοχές που ικανοποιούν αυτή τη συνθήκη ονομάζονται HSP (High-scoring Segment Pair). Η παράμετρος "T" καθορίζει την ταχύτητα και την ευαισθησία της αναζήτησης (Εικόνα 1). Ενώ το BLAST υπολογίζει τις παραμέτρους της EVD (Extreme Value Distribution), απο τις οποίες θα υπολογίσει τη στατιστική σημαντικότητα από εξομοιώσεις που έχει πραγματοποιήσει από πριν, το FASTA τις υπολογίζει από όλες τις άλλες ακολουθίες της βάσης δεδομένων και για αυτό το λόγο είναι και πιο αργό.

Εικονα 1: Ο αλγόριθμος του BLAST
Ο αλγόριθμος BLAST είναι μια διαισθητική (ευριστική) μέθοδος αναζήτησης που αναζητάει λέξεις μήκους W (προκαθορισμένο = 3 στο blastp) που σημειώνουν score μεγαλύτερο απο ένα προκαθορισμένο όριο (Threshold-T) όταν στοιχίζονται με την δοθείσα ακολουθία (query) και με ένα δεδομένο πίνακα υποκατάστασης. Οι λέξεις στη βάση δεδομένων που σημειώνουν score επεκτείνονται και προς τις δυο κατευθύνσεις σε μία προσπάθεια να μεγιστοποιηθεί το score, σύμφωνα με κάποιο κριτήριο ευαισθησίας (επίσης προκαθορισμένο απο πριν, συνήθως 10) του Ε-value. Στη συγκεκριμμένη περίπτωση η τριπλέτα PQG έχει βρεθεί να στοιχίζεται στην PMG, και καθώς χρησιμοποιείται ο πίνακας BLOSUM62, το score της στοίχισης είναι 13>Τ (P-P =7, G-G=6, Q-M=0). Κατόπιν αυτή η τριπλέτα θα επεκταθεί και προς τις δυο κατευθύνσεις για να δώσει τη μέγιστη τοπική στοίχιση. Τα HSPs που πληρούν αυτά τα κριτήρια θα αναφερθούν από το BLAST, υπό τον όρο ότι δεν υπερβαίνουν τoν αριθμό των επιτρεπτών προς εμφάνιση ακολουθιών.
Η περιοχή αναζήτησης ('Search' box) του BLAST δέχεται διαφορετικούς τύπους μορφοποιημένης εισόδου και αυτόματα καθορίζει τη μορφή. Οι τρεις μορφοποήσεις (formats) με τις οποίες μπορεί να εισαχθεί μια ακολουθία και να γίνει η αναζήτηση είναι:
Η ακολουθία σε μορφή FASTA ξεκινάει με μια γραμμή περιγραφής (single-line description), και ακολουθείται από γραμμές ακολουθίας με 60 χαρακτηρες για τα αμινοξέα σε κάθε γραμμή. Η γραμμή περιγραφής ξεχωρίζει από τις γραμμές της ακολουθίας επειδή στην αρχή της γραμμής περιγραφής υπάρχει το σύμβολο (">"). Ένα παράδειγμα ακολουθίας σε μορφή FASTA είναι:
>gi|129295|sp|P01013|OVAX_CHICK GENE X PROTEIN (OVALBUMIN-RELATED)
QIKDLLVSSSTDLDTTLVLVNAIYFKGMWKTAFNAEDTREMPFHVTKQESKPVQMMCMNNSFNVATLPA
EKMKILELPFASGDLSMLVLLPDEVSDLERIEKTINFEKLTEWTNPNTMEKRRVKVYLPQMKIEEKYNL
TSVMALGMTDLFIPSANLTGISSAESLKISQAVHGAFMELSEDGIEMAGSTGVIEDIKHSPESEQFRAD
HPFLFLIKHNPTNTIVYFGRYWSP
Εισάγωντας μια ακολουθία σε μορφή FASTA δεν επιτρέπεται να υπάρχουν άδειες γραμμές ενδιάμεσα. Οι ακολουθίες παρουσιάζονται με τους πρότυπους αμινοξικούς και νουκλεοτιδικούς κώδικες (IUB/IUPAC), με μερικές εξαιρέσεις. Ο κώδικας γραμμάτων (letter codes) των νουκλεοτιδίων είναι:
A --> adenosine M --> A C (amino) C --> cytidine S --> G C (strong) G --> guanine W --> A T (weak) T --> thymidine B --> G T C U --> uridine D --> G A T R --> G A (purine) H --> A C T Y --> T C (pyrimidine) V --> G C A K --> G T (keto) N --> A G C T (any) - --> gap of indeterminate length
Για τα προγράμματα που πραγματοποιούν αναζητήσεις αμινοξικών ακολουθιών ο κώδικας γραμμάτων των αμινοξέων ειναι:
A alanine P proline
B aspartate or asparagine Q glutamine
C cystine R arginine
D aspartate S serine
E glutamate T threonine
F phenylalanine U selenocysteine
G glycine V valine
H histidine W tryptophan
I isoleucine Y tyrosine
K lysine Z glutamate or glutamine
L leucine X any
M methionine * translation stop
N asparagine - gap of indeterminate
length
Σκέτη ακολουθία (Bare Sequence)
Είναι μόνο οι γραμμές της ακολουθίας χωρίς τη FASTA γραμμή ορισμού.
Προσδιοριστές (Identifiers)
Είναι ένας αριθμός πρόσβασης (accession) στο NCBI. Αυτοί οι προσδιοριστές ακολουθιών του NCBI έχουν μια συγκεκριμένη σύνταξη που περιγράφεται μέσα στη βάση ( ftp://ftp.ncbi.nlm.nih.gov/blast/documents/blastdb.txt )
Υπάρχουν πολλές παραλλαγές του προγράμματος BLAST, οι οποίες εκτελούν διαφορετικές εργασίες.
blastn - συγκρίνει μια νουκλεοτιδική ακολουθία (DNA) με μια βάση νουκλεοτιδικών ακολουθιών (DNA). Η αναζήτηση γίνεται και στις δύο αλυσίδες. Είναι ένα πρόγραμμα βελτιστοποιημένης ταχύτητας, όχι όμως και ευαισθησίας.
blastp - συγκρίνει την ζητούμενη αμινοξική ακολουθία με μια βάση πρωτεϊνικών ακολουθιών (σύγκριση πρωτεΐνης με πρωτεΐνες).
blastx - συγκρίνει μια άγνωστη νουκλεοτιδική ακολουθία (DNA) μεταφρασμένη σε όλα τα πλαίσια ανάγνωσης (reading frames) με μια βάση πρωτεϊνικών ακολουθιών του NCBI. Χρησιμοποιείται για την έυρεση πιθανών μεταφρασμένων πρωτεϊνικών προϊόντων μιας άγνωστης νουκλεοτιδικής ακολουθίας.
tblastn - συγκρίνει την ζητούμενη πρωτεϊνική ακολουθία με μια βάση νουκλεοτιδικών ακολουθιών (DNA) του NCBI που μεταφράζεται δυναμικά σε όλα τα πλαίσια ανάγνωσης (reading frames).
tblastx - μετατρέπει μια νουκλεοτιδική ακολουθία (DNA) σε μια πρωτεϊνική ακολουθία σε όλα τα πλαίσια ανάγνωσης (reading frames) και μετά τη συγκρίνει με μια βάση νουκλεοτιδικών ακολουθιών του NCBI η οποία έχει μεταφραστεί σε όλα τα πλαίσια ανάγνωσης (reading frames).
BLAST2 - Ονομάζεται εξελιγμένο (advanced) BLAST. Εκτελεί στοιχίσεις που περιέχουν κενά (gapped alignments).
MEGABLAST - είναι ένα πρόγραμμα που χρησιμοποιεί έναν πλεονεκτικό ("greedy algorithm") αλγόριθμο (Miller et al, 2000), για αναζήτηση στοίχισης νουκλεοτιδικών ακολουθιών. Χρησημοποιείται για στοίχιση ακολουθιών με μικρές διαφορές και είναι 10 φορές γρηγορότερο από παρόμοια προγράμματα. Ενδείκνυται για σύγκριση μεταξύ μεγάλων ακολουθιών.
PSI-BLAST - (Position Specific Iterated BLAST) χρησιμοποιεί σταθερή αναζήτηση, στην οποία οι ακολουθίες που θα βρεθούν στον πρώτο γύρο αναζητήσεων χρησιμοποιούνται για να χτίσουν ένα αποτελεσματικό μοντέλο για τους επόμενους κύκλους αναζητήσεων.
PHI-BLAST - (Pattern Hit Initiated BLAST) συνδυάζει το ταίριασμα ενός πρότυπου φυσιολογικής έκφρασης με μια συγκεκριμένη θέση που επαναλαμβάνεται στην πρωτεϊνική ακολουθία.
RPS-BLAST - συγκρίνει μια πρωτεϊνική ακολουθία ως προς την βάση Conserved Domain Database (CD-Search).
Αναλυτικό TUTORIAL για το BLAST υπάρχει στην διεύθυνση:
http://www.ncbi.nlm.nih.gov/BLAST/tutorial/Altschul-1.html
Το CLUSTALW είναι το πιο διαδεδομένο πρόγραμμα πολλαπλής στοίχισης βιολογικών ακολουθιών. Το πρόγραμμα χρησιμοποιεί έναν ιδιαίτερα πολύπλοκο αλγόριθμο προδευτικής στοίχισης (progressive alignment) για να κάνει σταδιακή στοίχιση πολλαπλών πρωτεϊνικών ή νουκλεοτιδικών (DNA) ακολουθιών. Όλες οι ακολουθίες πρέπει να είναι ένα αρχείο, η μία μετά την άλλη. Το πρόγραμμα CLUSTALW βρίσκεται στο EBI (European Bioinformatics Institute) ( http://www.ebi.ac.uk/clustalw/ ).
Το CLUSTALX (Thompson et al., 1994) είναι μια έκδοση του προγράμματος, το οποίο χρησιμοποιεί το περιβάλλον των X-Windows για να τρέχει το πρόγραμμα CLUSTALW. Το CLUSTALX δίνει την επιλογή δύο τρόπων στοίχισης ακολουθιών. Την πολλαπλή στοίχιση (multiple alignment mode) και την στοίχιση με βάση κάποιο profile (profile alignment mode), στη διεύθυνση http://www.ebi.ac.uk/clustalw/help.html ).
Μετά το τέλος των στοιχιζόμενων ακολουθιών επακολουθεί μια σειρά που μπορεί να περιέχει τους παρακάτω χαρακτήρες, η σημασία των οποίων είναι η εξής:
* = αυτή η στήλη της στοίχισης περιέχει ίδιο το αμινοξικό κατάλειπο σε όλες τις ακολουθίες (ή όμοιες βάσεις αν στοιχίζονται ακολουθίες DNA)
: = αυτή η στήλη της στοίχισης περιέχει διαφορετικά αλλά υψηλά διατηρημένα (παρόμοια) αμινοξέα.
. = αυτή η στήλη της στοίχισης περιέχει διαφορετικά αμινοξέα τα οποία είναι κατα κάποιον τρόπο όμοια.
= αυτή η στήλη της στοίχισης περιέχει ανόμοια αμινοξέα ή κενά (διαφορετικές βάσεις σε στοίχιση ακολουθιών DNA)
Τα φυλογενετικά δέντρα (Phylogenetic trees) μπορούν να υπολογιστούν από παλαιότερες στοιχίσεις ή μετά από μια πολλαπλή στοίχιση ενώ η στοίχιση βρίσκεται ακόμα σε εξέλιξη. Το CLUSTAL μπορεί να εκτελέσει πολλαπλές στοιχίσεις για περισσότερες απο 100 νουκλεοτιδικές (DNA) ή πρωτεϊνικές ακολουθίες αποτελούμενες μέχρι και 5000 κατάλοιπα (περιλαμβανομένων των κενών στην τελική στοίχιση).
Altschul, S.F., Gish, W., Miller, W., Myers. E.W., Lipman, D.J. (1990) Basic local alignment search tool, J Mol Biol., 215(3), 403-10
Altschul, S. F. and Gish, W. (1996) Local alignment statistics, Methods in Enzymology, 266, 460-480
Altschul, S. F., Madden, T. L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W. and Lipman, D.J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs, Nucleic Acids Res., 25, 3389-3402
Arratia, R. and Waterman, M. S. (1994) A phase transition for the score in matching random sequences allowing deletions Ann. Appl. Probab. 4: 200-225
Miller, W., Zhang, Z., Schwartz, S., Wagner, L. (2000) A greedy algorithm for aligning DNA sequences, Comput Biol, 7(1-2), 203-14
Pearson, W.R. (1990) Rapid and Sensitive Sequence Comparison with FASTP and FASTA, Methods in Enzymology, 183, 63- 98
Pearson, W.R. (1991) Searching protein sequence libraries: comparison of the sensitivity and selectivity of the Smith-Waterman and FASTA algorithms, Genomics, 11(3), 635-50
Pearson, W.R., Lipman, D.J. (1988) Improved tools for biological sequence comparison, Proc Natl Acad Sci., 85(8), 2444-8
Pearson, W. R. and Wood, T.C. (2001) Statistical significance in biological sequence comparison. In Handbook of Statistical Genetics. Balding, D.J, Bishop M., Cannings C. (eds). John Wiley and Sons, Ltd. England: 39-65
Smith, T.F., and Waterman, M.S. (1981) Identification of common molecular subsequences, J Mol Biol, 147(1), 195-7
Thompson, J.D., Higgins, D.G., Gibson, T.J. (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice, Nucleic Acids Res., 22, 4673-4680