ΒΙΟΠΛΗΡΟΦΟΡΙΚΗ
ΕΡΓΑΣΤΗΡΙΑΚΗ ΑΣΚΗΣΗ
HOMOLOGY MODELLING
Θεόδωρος Δ. Λιακόπουλος & Σταύρος Ι. Χαμόδρακας
Tομέας Βιολογίας Κυττάρου και Βιοφυσικής
Τμήμα Βιολογίας
Παν/μιο Αθηνών
Φεβρουάριος 2002
ΒΙΟΠΛΗΡΟΦΟΡΙΚΗ
ΕΡΓΑΣΤΗΡΙΑΚΗ ΑΣΚΗΣΗ
Φεβρουάριος 2002
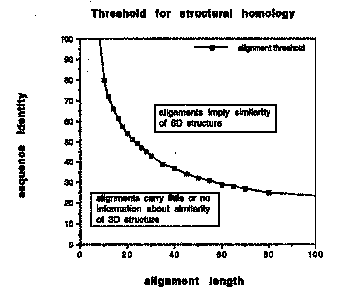
Με την προσέγγιση του Homology Modelling προσπαθούμε να προσδιορίσουμε θεωρητικά την άγνωστη δομή μιας πρωτεΐνης, εφόσον υπάρχει σημαντική ομοιότητα με μια πρωτεΐνη γνωστής δομής. Σε γενικές γραμμές, πρωτεΐνες με ομοιότητα στην ακολουθία άνω του 30%, η οποία εκτείνεται σε μήκος μεγαλύτερο των 80 καταλοίπων, θεωρείται πως διπλώνουν στο χώρο με παρόμοιο τρόπο. Τουλάχιστον αυτό ισχύει για τις περιοχές συγκεκριμένης (μη τυχαίας) δευτεροταγούς δομής. Η γνωστή δομή λέγεται Template (οδηγός), ενώ η άγνωστη Model (μοντέλο).
Η αναλυτική διαδικασία του Homology Modelling έχει ως εξής:
Η επιλογή του κατάλληλου οδηγού είναι σημαντική για την επιτυχία της προσπάθειας. Η αρχική αναζήτηση γίνεται με κάποιο πρόγραμμα εύρεσης ομοιοτήτων όπως το BLAST, έναντι μιας βάσης πρωτεϊνικών ακολουθιών γνωστής δομής. Είναι χαρακτηριστικό, ωστόσο, ότι η γνωστή δομή μπορεί να περιέχει λάθη. Έλεγχος της αξιοπιστίας μιας δομής, μπορεί να γίνει με προγράμματα όπως το WHAT_CHECK. (Αποτελέσματα του προγράμματος αυτού μπορούν να αναζητηθούν στη βάση δεδομένων PDBREPORT, για κάθε δομή κατατεθειμένη στην Protein DataBank.)
Η στοίχιση των ακολουθιών μοντέλου και οδηγού είναι καθοριστική για την επιτυχία του μοντέλου. Στοιχίσεις γίνονται με το χέρι, ή με τη βοήθεια λογισμικού όπως το καθιερωμένο CLUSTAL. Είναι σημαντικό, πως οι στοιχίσεις απεικονίζουν καταρχήν τη φυλογενετική συσχέτιση μεταξύ δύο αμινοξικών ακολουθιών, οι οποία δεν ταυτίζεται κατ' ανάγκη με τη δομική συσχέτιση. Για το λόγο αυτό, επιβάλλεται μερικές φορές η στοίχιση να τροποποιείται στα σημεία εισδοχών/διαγραφών (δηλαδή στα κενά - gaps), ώστε να είναι δυνατή η δημιουργία ρεαλιστικού μοντέλου. Η πρόγνωση δευτεροταγούς δομής στην ακολουθία του μοντέλου, μπορεί να μας δώσει επιπλέον βοήθεια για να τελειοποιήσουμε τη στοίχιση, ταιριάζοντας τα στοιχεία δευτεροταγούς δομής της πρόγνωσης με αυτά της πρωτεΐνης-οδηγού.
Η οπτική παρατήρηση του μοντέλου γίνεται με προγράμματα μοριακών γραφικών. Έτσι, μπορούμε να περιστρέψουμε τη δομή στο χώρο, καθώς και να συγκρίνουμε δύο δομές αντιπαραβάλλοντάς τις. Τα προγράμματα μπορεί επιπλέον να παρέχουν τη δυνατότητα τρισδιάστατης απεικόνισης (stereo) με χρήση ειδικών γυαλιών. Εργαλεία μοριακών γραφικών περιέχονται στα πακέτα WHAT IF, O, FRODO, τα εμπορικά Quanta/Insight και Sybyl, κ.ά. Το απλούστερο εργαλείο του είδους, ιδιαίτερα όμως πρακτικό και εύχρηστο, είναι το Rasmol/Chime.
Η βελτιστοποίηση ενός μοντέλου γίνεται με μεθόδους ενεργειακών υπολογισμών. Στην απλούστερη περίπτωση, κάνουμε ενεργειακή ελαχιστοποίηση του μοντέλου, με αποτέλεσμα να το φέρουμε στο πλησιέστερο ενεργειακό ελάχιστο. Σε άλλες περιπτώσεις, χρησιμοποιούμε αλγορίθμους που υλοποιούν τεχνικές Molecular Dynamics. Εκεί, κάνοντας προσομοίωση των αλληλεπιδράσεων μεταξύ όλων των ατόμων της δομής, έχουμε τη δυνατότητα να "διαταράξουμε" το μοντέλο, και να το οδηγήσουμε σε ένα περισσότερο απομακρυσμένο ενεργειακό ελάχιστο.
Θα προσπαθήσουμε να φτιάξουμε ένα μοντέλο για το ένζυμο διυδροφολική αναγωγάση του βακτηρίου Lactobacillus casei. Ως οδηγό θα χρησιμοποιήσουμε την λυμένη πειραματικά δομή του ίδιου ενζύμου του ανθρώπου. Η ακολουθία του ανθρώπινου ενζύμου έχει μήκος 186 κατάλοιπα, έναντι 162 του βακτηριακού. Αναμένουμε, άρα, να τοποθετηθούν κενά στην δεύτερη, κατά τη στοίχισή της με την πρώτη.
Ανασύρουμε από τη βάση SwissProt τις ακολουθίες των δύο πρωτεϊνών. Χρησιμοποιούμε το σύστημα SRS . Επιλέγουμε Start στην εισαγωγική σελίδα, "τσεκάρουμε" το μικρό τετράγωνο αριστερά από το όνομα της SwissProt, και επιλέγουμε Query forms>Standard. Στη φόρμα αναζήτησης, τοποθετούμε τους κωδικούς (IDs) των δύο πρωτεϊνών σε δύο από τα ελεύθερα κουτιά. Οι κωδικοί είναι DYR_LACCA και DYR_HUMAN. Επιλέγουμε αριστερά από τα κουτιά αυτά η αναζήτηση να γίνει στο πεδίο ID. Επιλέγουμε, τέλος, Combine searches with>OR. Το αποτέλεσμα είναι, να αναζητηθούν πρωτεϊνες που περιέχουν το ένα ή το άλλο ID, δηλαδή οι δύο πρωτεϊνες που μας ενδιαφέρουν. Δίνουμε την εντολή Submit Query. Αν όντως βρέθηκαν οι δύο πρωτεϊνες, μπορούμε να επιλέξουμε Perform operation on>Αll but selected, και στη συνέχεια Save. Θέλουμε, δηλαδή, να σώσουμε και τις δύο καταχωρήσεις που βρήκαμε (αφού δεν έχουμε επιλέξει καμία). Τελικά, επιλέγουμε Use view>FastaSeqs, και ξανά Save. Έχουμε έτσι στην οθόνη μας ένα αρχείο κειμένου που περιέχει τις ακολουθίες των δύο πρωτεϊνών σε μορφοποίηση Fasta. Το αρχείο αυτό, είναι κατάλληλο για να το εισάγουμε σε ένα πρόγραμμα στοίχισης. Έτσι, διαλέγουμε File>Save από την οριζόντια μπάρα του web browser, στη συνέχεια Save file as>Text, δίνουμε ένα όνομα και μια τοποθεσία για το αρχείο, και τελικά Save.
Στη συνέχεια, θα χρησιμοποιήσουμε για τη στοίχιση το πρόγραμμα CLUSTAL. Επιλέγουμε Output format>PIR, επειδή τη μορφοποίηση αυτή απαιτούν συνήθως τα προγράμματα homology modelling. Στη συνέχεια, εισάγουμε τις δύο ακολουθίες, αντιγράφοντάς τις στην περιοχή κειμένου από το αρχείο μας, ή επιλέγοντας Upload a file>Browse. Δίνουμε την εντολή Run CLUSTALW. Βλέπουμε έτσι τα αποτελέσματα της στοίχισης. Παρατηρούμε πως στην ακολουθία της πρωτεϊνης-οδηγού δεν υπάρχουν κενά, και υποθέτουμε έτσι ότι η δομή της θα είναι επαρκής για τη δημιουργία του μοντέλου. Στη συνέχεια, επιλέγουμε να δούμε το αρχείο με τη στοίχιση, πατώντας στο όνομα αρχείου με κατάληξη .aln. Τελικά, File>Save από την οριζόντια μπάρα του web browser, στη συνέχεια Save file as>Text, δίνουμε ένα όνομα και μια τοποθεσία για το αρχείο, και Save. Από το αρχείο αυτό, χρησιμοποιούμε έναν συντάκτη κειμένου όπως το Notepad για να δημιουργήσουμε δύο αρχεία τελικά, το καθένα από τα οποία θα περιέχει και μία ακολουθία. Οι ακολουθίες πρέπει να παραμείνουν σε μορφοποίηση PIR, με τις δύο εισαγωγικές γραμμές. ενώ η ακολουθία της βακτηριακής πρωτεϊνης περιέχει φυσικά και τα κενά (-) της στοίχισης.
Το επόμενο βήμα είναι να εντοπίσουμε την λυμένη πειραματικά δομή (οδηγό), η οποία βρίσκεται στην Protein DataBank. Εκεί βλέπουμε ένα πλαίσιο αναζήτησης στην PDB, όπου γράφουμε τον κωδικό 1DHF, ο οποίος αντιστοιχεί σε μία από τις προσδιορισμένες δομές της πρωτεϊνης μας. "Τσεκάρουμε" την επιλογή Query by ID only, αφού γνωρίζουμε τον κωδικό (ID), και επιλέγουμε Find a structure. Βλέπουμε περιληπτικές πληροφορίες για τη δομή αυτή, και επιλέγουμε Download/display file. Στη συνέχεια Download the structure file, αφού χρειαζόμαστε σε αρχείο τις συντεταγμένες τις δομής-οδηγού για το homology modelling. Κάνουμε δεξί κλικ στο X που αντιστοιχεί στη στήλη File format>PDB και στη γραμμή Compression>None. Επιλέγουμε Save target as... και σώζουμε και αυτό το αρχείο.
Έχουμε τώρα ό,τι χρειάζεται για να προχωρήσουμε στο homology modelling. Θα χρησιμοποιήσουμε ένα δικτυακό τόπο που προσφέρει πρόσβαση στο πρόγραμμα WHAT IF. Επιλέγουμε Server classes>Βuild/check/repair model, και στη συνέχεια Homology modelling. Στη φόρμα που εμφανίζεται, εισάγουμε διαδοχικά με τα πλήκτρα Browse το αρχείο με τη δομή οδηγό, το αρχείο με την ακολουθία που αντιστοιχεί στη δομή αυτή, και τέλος το αρχείο με την ακολουθία της "άγνωστης" πρωτεϊνης, το οποίο περιέχει και τα κενά της στοίχισης. Επιλέγουμε Send. Όταν το πρόγραμμα εκτελεστεί, επιλέγουμε Results. Κάνουμε δεξί κλικ στο αρχείο model.pdb, το οποίο περιέχει τις συντεταγμένες του μοντέλου, και το σώζουμε.
Το μοντέλο μπορούμε να το παρατηρήσουμε οπτικά με κάποιο πρόγραμμα μοριακών γραφικών. Επίσης, μπορούμε να το συγκρίνουμε με κάποια άλλη δομή, ταιριάζοντάς το στο χώρο με αυτήν. Η δομή της βακτηριακής πρωτεϊνης έχει πράγματι προσδιοριστεί, και μπορούμε να την αναζητήσουμε στην PDB (όπως παραπάνω) με τον κωδικό 3DFR. Έτσι, αφού σώσουμε το αντίστοιχο αρχείο, επιλέγουμε στη σελίδα του WHAT IF Βuild/check/repair model και Compare A Model To A Solved Structure. Στέλνουμε στο πρόγραμμα αυτό το μοντέλο μας, καθώς και την λυμένη δομή (στο πεδίο Template). Επιλέγουμε Results, και στα αναλυτικά αποτελέσματα που παρουσιάζονται εντοπίζουμε το πεδίο All residues. Εκεί μπορούμε να δούμε την παράμετρο RMS συνολικά για όλα τα κατάλοιπα, που είναι μέτρο της απόκλισης μεταξύ των δομών που συγκρίνονται. Παρατηρούμε ειδικά το RMS για τα άτομα α-άνθρακα (Alpha carbons), καθώς και για τα κέντρα των καταλοίπων (Residue centers). Με βάση τις παραμέτρους αυτές, είναι δυνατό να πιθανολογήσουμε για την επιτυχία του modelling.
Στο διαδίκτυο μπορεί να χρησιμοποιήσει κανείς προγράμματα που εκτελούν αυτόματα homology modelling, με μόνο δεδομένο από την πλευρά του χρήστη την ακολουθία της άγνωστης πρωτεϊνης. Τα προγράμματα αυτά αναλαμβάνουν την αναζήτηση κατάλληλης δομής-οδηγού, τη στοίχιση, καθώς και τη δημιουργία του μοντέλου. Δεν υπάρχει δυνατότητα παρέμβασης του χρήστη στη διαδικασία, μπορεί όμως αυτός να κάνει στο τέλος επισκόπηση των βημάτων που ακολουθήθηκαν αυτόματα. Ένα τέτοιο πρόγραμμα είναι το Swiss Model, που χρησιμοποιείται από τη διεύθυνση http://www.expasy.ch/swissmod/SWISS-MODEL.html. Τα αποτελέσματα αποστέλλονται στο χρήστη μέσω e-mail.
Με βάση τα πρωτόκολλα που αναφέραμε, προσπαθήστε να δημιουργήσετε ένα μοντέλο για την πρωτεϊνη άγνωστης δομής DYR_MESAU (SwissProt ID).