ΒΙΟΦΥΣΙΚΗ
ΕΡΓΑΣΤΗΡΙΑΚΗ ΑΣΚΗΣΗ
ΠΡΟΒΛΕΨΗ ΤΗΣ ΔΕΥΤΕΡΟΤΑΓΟΥΣ ΔΟΜΗΣ ΜΙΑΣ ΠΡΩΤΕΪΝΗΣ
ΑΠΟ ΤΗΝ ΑΜΙΝΟΞΙΚΗ ΤΗΣ ΑΚΟΛΟΥΘΙΑ
Εφαρμογή στην πρόβλεψη της δευτεροταγούς δομής της
πρωτεΐνης bovine pancreatic trypsin inhibitor (BPTI)
Βασιλική Α. Οικονομίδου & Σταύρος Ι. Χαμόδρακας
Tομέας Βιολογίας Κυττάρου και Βιοφυσικής
Τμήμα Βιολογίας
Παν/μιο Αθηνών
Φεβρουάριος 2002
Για να καταλάβουμε τη 'λογική' στην οποία στηρίζεται η βιολογική δράση μιας πρωτεΐνης είναι αναγκαία η γνώση της στερεοδιάταξής της.
Προς το παρόν, μόνο μια λεπτομερής κρυσταλλογραφική ανάλυση με περίθλαση ακτίνων - Χ (μια μέθοδος εξαιρετικά κοπιαστική, που παίρνει πολύ χρόνο και είναι εξαιρετικά δαπανηρή) μπορεί να δώσει πληροφορίες για τη στερεοδιάταξη μιας πρωτεΐνης, με την προϋπόθεση ότι κατάλληλοι κρύσταλλοι της πρωτεΐνης μπορούν να αναπτυχθούν. Ήδη οι τεχνικές της κρυσταλλογραφίας ακτίνων-Χ έχουν προσδιορίσει μ' επιτυχία πάνω από 10.000 πρωτεϊνικές δομές (Αναφ. 1,2). Επίσης, η φασματοσκοπία NMR.
Αφού η φυσική στερεοδιάταξη μιας πρωτεΐνης είναι κωδικοποιημένη στην αμινοξική της ακολουθία (αλληλουχία καταλοίπων της), όπως δείχθηκε αρκετά πειστικά από πειράματα αναδιάταξης (3), έγιναν πολλές προσπάθειες να προβλεφθεί η πρωτεϊνική δομή από την αμινοξική της ακολουθία.
Στις αρχικές μελέτες χρησιμοποιήθηκαν φυσικοχημικές μέθοδοι (π.χ. οπτικός στροφικός διασκεδασμός, O.R.D) για να συσχετιστεί η αμινοξική σύσταση και η περιεκτικότητα των πρωτεϊνών σε έλικες.
Kατόπιν πιστεύθηκε ότι θα μπορούσαμε να εφαρμόσουμε τις αρχές της στερεοδιαταξικής ανάλυσης που αναπτύχθηκαν για μικρά μόρια (βλ. Ασκ. 1,2) στον προσδιορισμό της στερεοδιάταξης μιας πρωτεΐνης.
Μ’ αυτές τις μεθόδους βρίσκονται οι σταθερές στερεοδιατάξεις ενός μορίου, μεταβάλοντας εκλεγμένες μεταβλητές ώστε να ελαχιστοποιηθεί η στερεοδιαταξική ενέργεια, που εκφράζεται σαν αναλυτική συνάρτηση των ατομικών θέσεων, των χημικών δεσμών και των ενδοατομικών δυνάμεων.
Η εφαρμογή τέτοιων μεθόδων δεν έχει δώσει ακόμη ικανοποιητικά αποτελέσματα, γιατί υπάρχουν αρκετά προβλήματα να ξεπεραστούν.
Συγκεκριμένα:
> Oι πρωτεΐνες έχουν πολλά άτομα. Επομένως οι υπολογισμοί της ενέργειας παίρνουν πολύ χρόνο, αφού η ενέργεια υπολογίζεται σαν το άθροισμα των αλληλεπιδράσεων μεταξύ ζευγαριών ατόμων.
> Οι πρωτεΐνες είναι σταθερές στο Η2Ο σε θερμοκρασία περιβάλλοντος. Ενώ οι στερεοδιαταξικές ενέργειες μικρών μορίων, in vacuo, και σε χαμηλές θερμοκρασίες μπορούν να υπολογιστούν εύκολα, πολύ λίγα είναι γνωστά για τα αποτελέσματα του διαλύτη και της ατομικής θερμικής κίνησης στις ενδοατομικές δυνάμεις.
> Οι πρωτεϊνικές δομές περιγράφονται χρησιμοποιώντας πολλές μεταβλητές. Ακόμη και στην περίπτωση που θα θεωρήσουμε ως μεταβλητές τις γωνίες στροφής γύρω από απλούς δεσμούς, μια μικρή πρωτεΐνη για να περιγραφεί μόνο χρειάζεται μερικές εκατοντάδες βαθμούς ελευθερίας (γωνίες στροφής), πράγμα που κάνει την ελαχιστοποίηση της ενέργειας πολύ δύσκολη.
> Στους υπολογισμούς της ενέργειας εμφανίζονται πολλά ελάχιστα κι αυτό κάνει δύσκολη την εκλογή του σωστού ελαχίστου που αντιπροσωπεύει τη φυσική στερεοδομή.
To πιο ενοχλητικό απ΄αυτά τα προβλήματα είναι ίσως η ύπαρξη πολλών ελαχίστων ενέργειας στους υπολογισμούς.
Για να παρακάμψουμε τη δυσκολία που υπεισάγει η ύπαρξη πολλών ελαχίστων ενέργειας, έχουν επινοηθεί διάφοροι εμπειρικοί αλγόριθμοι πρόβλεψης δευτεροταγούς δομής μιας πρωτεΐνης. Η ιδέα είναι να μπορέσουμε να πάρουμε μια ‘αρχική’ στερεοδιάταξη, από την οποία ξεκινώντας την ελαχιστοποίηση της ενέργειας να οδηγηθούμε στη φυσική στερεοδομή.
Από τις μεθόδους αυτές ενδεικτικά αναφέρουμε των Chou και Fasman, Nagano, Lim, Scheraga και συνεργατών, Robson και συνεργατών, Ptitsyn και Finkelstein, Kabat και Wu, Dufton και Hider.
Τα τελευταία χρόνια έχουν αναπτυχθεί πολλές άλλες 'δημοφιλείς' μέθοδοι πρόβλεψης δευτεροταγούς δομής, από τις οποίες αναφέρουμε τις εξής:
To PHDsec προβλέπει τη δευτεροταγή δομή μιας πρωτεϊνης με πολλαπλή στοίχιση ακολουθιών. Η δευτεροταγής δομή προβλέπεται από ένα σύστημα νευρωνικών δικτύων με επιτυχία που κατά μέσο όρο είναι >72% για τρεις καταστάσεις: α-έλικα, β-πτυχωτές επιφάνειες και β-στροφές/τυχαία δομή. (Rost, 1996; Rost & Sander, 1994; Rost & Sander, 1993).
Οι προβλέψεις του PHDsec έχουν τρία κύρια χαρακτηριστικά:
> Βελτιωμένη επιτυχία μέσω εξελικτικής πληροφορίας από πολλαπλή στοίχιση ακολουθιών.
> Βελτιωμένη πρόβλεψη β-πτυχωτών επιφανειών μέσω μιας ισορροπημένης διαδικασίας μάθησης.
> Μεγαλύτερης ακρίβεια πρόβλεψης δευτεροταγούς δομής με τη χρήση πολυ-επιπεδου συστήματος.
Το PSIPRED (Jones, 1999) είναι μια απλή και αξιόπιστη μέθοδος πρόβλεψης δευτεροταγούς δομής ενσωματώνοντας δύο feed-forward νευρωνικά δίκτυα, τα οποία εκτελούν την ανάλυση σε αποτελέσματα που έχουν παρθεί από το PSI-BLAST (Position Specific Iterated - BLAST) (Altschul et al., 1997).
Η έκδοση 2.0 του PSIPRED (Jones, 1999) περιλαμβάνει ένα νέο αλγόριθμο που υπολογίζει το μέσο όρο των αποτελέσμάτων μέχρι και 4 νευρωνικών δικτύων στη διαδικασία της πρόβλεψης, αυξάνοντας την ακρίβεια της. Η αξιολόγηση της μεθόδου, κατά τον συγγραφέα, δίνει ένα ποσοστό επιτυχίας Q3 της τάξεως του 78%.
Το Jpred δεν είναι ένα μεμονωμένο πρόγραμμα αλλά ένα δικτυακό σύστημα που τρέχει μια ποικιλία προγραμμάτων πρόβλεψης δευετροταγούς δομής και συγκρίνει τα αποτελέσματά τους. Άρα είναι ένας server που δέχεται είτε μια πρωτεϊνική ακολουθία, είτε πολλαπλή στοίχιση πρωτεϊνικών ακουλουθιών και προβλέπει τη δευτεροταγή δομή. Δηλαδή, δουλεύει συνδυάζοντας έναν αριθμό μοντέρνων, υψηλής ποιότητας μεθόδων πρόβλεψης για να φτιάξει ένα συνδυαστικό αποτέλεσμα πρόβλεψης. (Cuff & Barton, 1999).
H σημασία των εμπειρικών αλγορίθμων πρόβλεψης, εκτός από το γεγονός ότι μπορούν να δώσουν μια 'αρχική στερεοδιάταξη' σε προβλήματα ελαχιστοποίησης της ενέργειας, περιορίζοντας έτσι το ψάξιμο για τη φυσική δομή, όπως προαναφέρθηκε, έγκειται στα εξής:
> Είναι πολύτιμοι σε περιπτώσεις που είναι αναγκαίο να βρούμε πώς 'διπλώνεται' η πολυπεπτιδική αλυσίδα σε χάρτες ηλεκρονικής πυκνότητας μικρής διακριτικότηταςστην κρυσταλλογραφία πρωτεϊνών.
> Οι στερεοδιαταξικές παράμετροι (θ' αναφερθούν παρακάτω) βοηθάνε πολλές φορές να βρεθούν περιοχές σε πρωτεΐνες, στις οποίες παρατηρούνται στερεοδιαταξικές αλλαγές που επάγονται είτε από αλλαγές στο περιβάλλον της πρωτεΐνης, είτε οφείλονται σε μεταλλαγές που προκαλούν μεταλλαγή της αμινοξικής ακολουθίας της (conformational switches).
> Πολλές φορές είναι χρήσιμοι στην αναγνώριση δομημένων περιοχών σε ομόλογες πρωτεΐνες.
> Μπορούν επίσης να εισηγηθούν λογική σχεδίαση συνθετικών αναλόγων για πειραματικές εξετάσεις, για να δούμε αν διατήρηση ή αλλαγές σε στερεοδιάταξη προκαλούν ή όχι διατήρηση ή απώλεια ενζυμικής δράσης.
Στην άσκηση, δεν θα επιχειρήσουμε να αναπτύξουμε όλες τις μεθόδους πρόγνωσης της δευτεροταγούς δομής πρωτεϊνών από την αμινοξική τους ακολουθία. Απλώς θα προσπαθήσουμε να αναλύσουμε περιληπτικά τη μέθοδο Dufton & Hider (6). H μέθοδος αυτή αποτελεί μια παραλλαγή της δημοφιλούς για την απλότητα και την ακρίβειά της μεθόδου, των Chou & Fasman (4,5).
Θα χρησιμοποιήσουμε τη μέθοδο των Dufton & Hider για την πρόβλεψη της δευτεροταγούς δομής της πρωτεΐνης bovine pancreatic trypsin inhibitor, της οποίας η κρυσταλλοδομή έχει προσδιοριστεί, και στη συνέχεια θα επιχειρήσουμε ν΄αξιολογήσουμε την ικανότητα πρόβλεψης της μεθόδου, για να βγάλουμε συμπεράσματα για το πόσο χρήσιμη μπορεί να φανεί η μέθοδος.
Η μέθοδος των Dufton και Hider
Συνοπτικά η μέθοδος των Dufton και Hider μπορεί να περιγραφεί ως εξής:
Από στατιστική ανάλυση, αρχικά 15 (αν. 4, 5) και κατόπιν 29 (αν. 7, 8) πρωτεϊνών με 4741 αμινοξικά κατάλοιπα συνολικά (οι δομές των οποίων έχουν προσδιοριστεί κρυσταλλογραφικά), οι Chou & Fasman απέδωσαν στα 20 κατάλοιπα στερεοδιαταξικές παραμέτρους, που εκφράζουν την ικανότητά τους να συμμετέχουν στον προσδιορισμό μιας ορισμένης δευετροταγούς δομής: α-έλικας, εκτεταμένης δομής (πιθανόν β-πτυχωτής επιφάνειας) ή στροφής.
Οι παράμετροι για ένα συγκεκριμένο κατάλοιπο συμβολίζονται με Pα, Pβ, Pτ, αντίστοιχα.
Ο ορισμός των παραμέτρων και ο τρόπος με τον οποίο υπολογίστηκαν από τους Chou & Fasman δείχνονται με το εξής υποθετικό παράδειγμα:
Aν υποθέσουμε ότι ένα συγκεκριμένο κατάλοιπο π.χ. η ΑLA, εμφανίζεται 400 φορές στο σύνολο των 4741 καταλοίπων έτσι ώστε 200 φορές να βρίσκεται σε δομή α-έλικας, 100 σ΄εκτεταμένη δομή, 50 σε β-στροφές και 50 σε ακανόνιστη δευτεροταγή δομή, τότε, η συχνότητα εμφάνισής της σε κάθε δομή είναι:
α-έλικες: fα=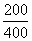 =0,5
=0,5
εκτεταμένη δομή: fβ=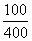 =0,25
=0,25
β-στροφές: fτ=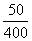 =0,125
=0,125
ακανόνιστη στερεοδιάταξη: fc==0,125
Με όμοιο τρόπο μπορούν να υπολογιστούν οι συχνότητες fα, fβ, fτ, fc για κάθε ένα από τα 20 αμινοξικά κατάλοιπα. Οι μέσες τιμές των fα, fβ, fτ υπολογίζονται από τις σχέσεις:
< fα > =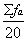
< fβ > =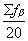
< fτ > =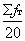
Οι Chou & Fasman όρισαν σαν στερεοδιαταξικές παραμέτρους για κάθε κατάλοιπο τις:
Pα =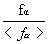
Pβ =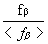
Pτ =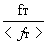
Τιμές των Pα, Pβ, Pτ δίνονται στον Πίνακα Ι και προέρχονται, όπως αναφέρθηκε παραπάνω, από τη στατιστική ανάλυση 29 πρωτεϊνών με 4741 κατάλοιπα συνολικά (αν. 7,8).
Ο σχηματισμός α-ελίκων, εκτεταμένων δομών (β-πτυχωτών επιφανειών) ή β-στροφών είναι συνεργατικό φαινόμενο, αφού για να σχηματιστεί μια δευτεροταγής δομή πρέπει διαδοχικά κατάλοιπα ν΄αποκτήσουν τη σωστή στεροδιάταξη. Έτσι, σε μια έλικα π.χ., για να επιτευχθούν αλληλεπιδράσεις που τη σταθεροποιούν, πρέπει τουλάχιστον 4 διαδοχικά κατάλοιπα να αποκτήσουν τη σωστή στερεοδομή. Υπολογισμοί στατιστικής μηχανικής δείχνουν ότι η δημιουργία μιας έλικας στηρίζεται στο σχηματισμό ενός ‘πυρήνα’ που ακολουθείται από ανάπτυξη της έλικας. Αυτό σημαίνει ότι τουλάχιστον 4 ή περισσότερα κατάλοιπα που ευνοούν το σχηματισμό ελίκων (δηλαδή με Pα>1.0) πρέπει να δημιουργήσουν ένα ‘πυρήνα’ για να αρχίσει μια α-έλικα. Η έλικα μπορεί να επεκταθεί (σε οποιαδήποτε διεύθυνση) με μια σειρά καταλοίπων που μπορεί να περιέχουν κατάλοιπα ευνοϊκά για το σχηματισμό ελίκων και πιθανόν και ένα μικρό ποσοστό αντιελικοειδών καταλοίπων, και τελειώνει με μια σειρά καταλοίπων που προτιμούν άλλη στερεοδιάταξη.
Όπως δείχθηκε θεωρητικά και πειραματικά (αν. 4,5), το 'κρίσιμο' μέγεθος για το σχηματισμό ελίκων, εκτεταμένων δομών και β-στροφών είναι αντίστοιχα 6, 5 και 4 κατάλοιπα.
Η διαδικασία που ακολουθείται στη μέθοδο των Dufton & Hider για τον προσδιορισμό της δευτεροταγούς δομής μιας πρωτεΐνης, κάνοντας χρήση των στερεοδιαταξικών παραμέτρων και της συνεργατικότητας που προαναφέρθηκε, είναι η εξής:
Γνωρίζοντας την αμινοξική ακολουθία της πρωτεΐνης ξεκινάμε την ανάλυση από το αμινοτελικό άκρο της (απ΄όπου αρχίζει η αρίθμηση των καταλοίπων). Αρχικά υπολογίζουμε το γινόμενο των στερεοδιαταξικών παραμέτρων για α-έλικα (Pα) των καταλοίπων 1, 2, 3, 4 5, 6, το γινόμενο των Pβ για τα κατάλοιπα 1,2,3,4,5 και το γινόμενο των Pτ για τα κατάλοιπα 1,2,3,4.
Έστω ότι τα γινόμενα είναι: Να1, Νβ1, ΝT1 αντίστοιχα.
Είναι φανερό από την προηγούμενη συζήτηση ότι, τα γινόμενα αυτά εκφράζουν την ικανότητα του πρώτου εξαπεπτιδίου, πενταπεπτιδίου και τετραπεπτιδίου αντίστοιχα, να δημιουργήσουν α-έλικα, εκτεταμένη δομή ή β-στροφή (αν Να1 ή Νβ1 ή ΝT1 > 1.0 ευνοείται η δημιουργία της αντίστοιχης δευτεροταγούς δομής).
Προχωράμε υπολογίζοντας το γινόμενο των Ρα για τα κατάλοιπα 2, 3, 4, 5, 6, 7 έστω Να2, των Ρβ γαι τα κατάλοιπα 2, 3, 4, 5, 6, έστω Νβ2 και των ΡΤ για τα κατάλοιπα 2, 3, 4, 5 έστω ΝΤ2 και συνεχίζουμε με τον ίδιο τρόπο μέχρι να εξαντληθούν όλα τα κατάλοιπα. Θα καταλήξουμε να έχουμε τρεις σειρές παραμέτρων Να1Να2...Ναn-5, Νβ1Νβ2...Νβn-4, ΝΤ1ΝΤ2...ΝΤn-3 (όπου n, ο αριθμός των καταλοίπων της πρωτεΐνης), που εκφράζουν την ικανότητα διαδοχικών εξα-, πεντα- και τετρα- πεπτιδίων να δημιουργήσουν α-έλικα, εκτεταμένη δομή ή β-στροφή. Η έκταση των περιοχών που παρουσιάζουν μια ορισμένη δευτεροταγή δομή βρίσκεται από την επικάλυψη ευνοϊκών εξα-, πεντα- και τετρα- πεπτιδίων, αντίστοιχα. Όταν δεν υπάρχει επικάλυψη, αν π.χ. δύο ευνοϊκά εξαπεπτίδια συναντιώνται άκρη-άκρη, αυτό σημαίνει ότι υπάρχει μια μικρή περιοχή χωρίς ορισμένη δευτεροταγή δομή.
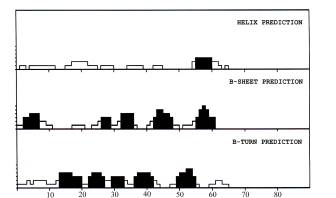
Τ' αποτελέσματα μπορούν να παρασταθούν γραφικά. Μ' αυτό τον τρόπο μπορούμε να έχουμε προφίλ κατά μήκος της αμινοξικής ακολουθίας της πρωτεΐνης που δείχνουν άμεσα τις δομημένες περιοχές της και το ποιά δομή ευνοείται. Η μεγαλύτερη δυσκολία που παρουσιάζεται στην ερμηνεία των αποτελεσμάτων, οφείλεται συνήθως στην ύπαρξη περιοχών στις οποίες ευνοείται ο σχηματισμός δομών περισσοτέρων από μια. Σε τέτοιες περιπτώσεις, επειδή άλλοι παράγοντες πιθανόν να ευνοούν το σχηματισμό της μιας ή της άλλης δομής, αναγκαζόμαστε συνήθως να παρουσιάζουμε τις προβλέψεις μας ξεχωριστά.
Πίνακας Ι
Στερεοδιαταξικές παράμετροι των 20 αμινοξικών καταλοίπων για α-έλικα, εκτεταμένη δομή και β-στροφή.
| Κατάλοιπο | Ρα | Ρβ** | ΡΤ1 | ΡΤ2 | ΡΤ3 | ΡΤ4 | ΡΤ | ΡΤΜ |
| GLY | 0.57 | 0.75 | 1.23 | 1.01 | 2.05 | 1.78 | 1.54 | 1.58 |
| ALA | 1.42 | 0.83 | 0.65 | 0.96 | 0.43 | 0.67 | 0.64 | 0.67 |
| VAL | 1.06 | 1.70 | 0.64 | 0.55 | 0.35 | 0.75 | 0.53 | 0.45 |
| LEU | 1.21 | 1.30 | 0.66 | 0.29 | 0.40 | 0.81 | 0.57 | 0.37 |
| ILE | 1.08 | 1.60 | 0.68 | 0.41 | 0.27 | 0.72 | 0.54 | 0.37 |
| PRO | 0.57 | 0.55 | 1.36 | 3.42 | 0.59 | 0.77 | 1.56 | 2.07 |
| PHE | 1.13 | 1.38 | 0.85 | 0.43 | 0.67 | 0.85 | 0.64 | 0.56 |
| TRP | 1.08 | 1.37 | 0.80 | 0.13 | 0.80 | 1.99 | 1.00 | 0.50 |
| ASR | 1.01 | 0.54 | 1.68 | 1.27 | 2.06 | 0.90 | 1.43 | 1.60 |
| GLU | 1.51 | 0.37 | 0.75 | 0.89 | 1.06 | 0.89 | 0.82 | 0.93 |
| TYR | 0.69 | 1.47 | 1.07 | 0.79 | 1.24 | 1.52 | 1.13 | 1.04 |
| ASN | 0.67 | 0.89 | 1.78 | 0.96 | 2.15 | 1.01 | 1.51 | 1.53 |
| GLN | 1.11 | 1.10 | 0.84 | 1.03 | 0.45 | 1.22 | 0.97 | 0.77 |
| THR | 0.83 | 1.19 | 0.95 | 1.22 | 0.81 | 0.93 | 0.99 | 1.00 |
| SER | 0.77 | 0.75 | 1.38 | 1.47 | 1.32 | 1.07 | 1.35 | 1.40 |
| CYS | 0.70 | 1.19 | 1.77 | 0.77 | 1.32 | 1.43 | 1.29 | 0.95 |
| MET | 1.45 | 1.05 | 0.86 | 0.86 | 0.14 | 0.72 | 0.65 | 0.47 |
| HIS | 1.00 | 0.87 | 1.53 | 0.48 | 1.13 | 0.56 | 0.94 | 0.78 |
| LYS | 1.16 | 0.74 | 0.54 | 1.35 | 0.81 | 1.02 | 0.95 | 1.12 |
| ARG | 0.98 | 0.93 | 0.73 | 1.31 | 1.39 | 0.80 | 1.05 | 1.22 |
**Μια β-στροφή αποτελείται απο 4 διαδοχικά κατάλοιπα (Αναφ. 7). Οι παράμετροι ΡΤ1, ΡΤ2, ΡΤ3, ΡΤ4 αναφέρονται στην 1η, 2η, 3η, 4η θέση της στροφής αντίστοιχα. ΡΤ και ΡΤΜ είναι μέσες τιμές των παραμέτρων λαμβάνοντας υπ’ όψη τις 4 θέσεις και τις 2 μεσαίες θέσεις της στροφής αντίστοιχα (Αναφ. 7).
ΠΕΙΡΑΜΑΤΙΚΟ ΜΕΡΟΣ
Στην άσκηση θα προβλέψουμε τη δευτεροταγή δομή της πρωτεΐνης bovine pancreatic trypsin inhibitor (BPTI) με τη μέθοδο των Dufton και Hider. Οι υπολογισμοί θα γίνουν με τη βοήθεια προγράμματος ηλεκτρονικού υπολογιστή (που γράφτηκε από το συγγραφέα) όπως περιγράφεται στο παράρτημα.
Το πρόγραμμα δέχεται σαν δεδομένο την αμινοξική ακολουθία μιας ή περισσοτέρων πρωτεϊνών και δίνει για κάθε πρωτεΐνη ως αποτελέσματα τις παραμέτρους Να1...Ναn-5, Νβ1...Νβn-4, ΝΤ1...ΝΤn-3 και επίσης προφίλ κατά μήκος της αμινοξικής ακολουθίας, ώστε να είναι δυνατός ο άμεσος προσδιορισμός και το είδος των δομημένων περιοχών.
Με το πρόγραμμα επίσης είναι δυνατόν να γίνει άμεση σύγκριση προγνωστικών αποτελεσμάτων και πειραματικών δεδομένων (εφ' όσον υπάρχουν) και εφαρμογή άλλων προγνωστικών μεθόδων.
Για να κρίνουμε το πόσο καλός ή όχι είναι ένας εμπειρικός αλγόριθμος πρόβλεψης, διάφοροι συγγραφείς πρότειναν τη χρήση διαφόρων 'δεικτών συμφωνίας' μεταξύ πειραματικών δεδομένων και προγνωστικών αποτελεσμάτων.
Ένας από τους δείκτες αυτούς είναι και ο Q δείκτης των Chou και Fasman (4, 5) που θα χρησιμοποιήσουμε στην άσκηση για την απλότητα του. Ο υπολογισμός του Q για μια συγκεκριμένη δομή έστω π.χ. α-έλικα, γίνεται ως εξής:
Έστω :
Ν=αριθμός καταλοίπων πρωτεΐνης που εξετάζουμε
w=κατάλοιπα που πρoβλέφθηκαν ελικοειδή και παρατηρήθηκαν ελικοειδή
x=κατάλοιπα που πρoβλέφθηκαν μη-ελικοειδή και παρατηρήθηκαν μη-ελικοειδή
y=κατάλοιπα που πρoβλέφθηκαν μη-ελικοειδή και παρατηρήθηκαν ελικοειδή
z=κατάλοιπα που πρoβλέφθηκαν ελικοειδή και παρατηρήθηκαν μη-ελικοειδή
τότε 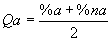
όπου 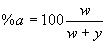
και 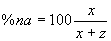
Αφού παρθούν τ' αποτελέσματα πρόβλεψης στην αμινοξική ακολουθία της bovine pancreatic trypsin inhibitor και μελετηθούν με τη βοήθεια του υπευθύνου, θα γίνει ο υπολογισμός των δεικτών Qα, Qβ και QΤ και θ' απαντηθεί (γραπτά) η ερώτηση:
Πώς, κατά τη γνώμη σας, θα μπορούσαμε να χρησιμοποιήσουμε τα προγνωστικά αποτελέσματα που πήραμε;
ΠΑΡΑΡΤΗΜΑ
Εκτέλεση του προγράμματος
Για την εκτέλεση του προγράμματος, που προβλέπει τη δευτεροταγή δομή της bovine pancreatic trypsin inhibitor με την μέθοδο των Dufton και Hider, πρέπει να δοθούν τα ακόλουθα δεδομένα στο πακέτο SECSTR (Hamodrakas, 1988):
TEST RUN [Τίτλος]
1 [Αριθμός αμινοξικών ακολουθιών που εξετάζονται]
PROTEIN TRYPSIN INHIBITOR [Όνομα πρωτεΐνης]
58 [Αριθμός αμινοξικών καταλοίπων της πρωτεΐνης]
RPDFCLEPPYTGPCKARIIRYFYNAKAGLCQTFVYGGCRAKRNNFKSAEDCMRTCGGA 1
*ΟΟΟΟΟΟ******////////////**//////////*****////ΟΟΟΟΟΟΟΟΟ*** 2
|
1[Αμινοξική ακολουθία της πρωτεΐνης. Τα αμινοξικά κατάλοιπα παριστάνονται μ' ένα κωδικό γράμμα το καθένα. Η πρωτεΐνη έχει 58 κατάλοιπα].
2[Η δευτεροταγής δομή της πρωτεΐνης όπως παρατηρήθηκε πειραματικά (βλ. Αναφ.9). Το σύμβολο Ο παριστάνει κατάλοιπο σε α-έλικα, το / σ' εκτεταμένη δομή (β-πτυχωτές επιφάνειες) και το * έλλειψη ορισμένης δευτεροταγούς δομής (random coil) ή β-στροφές (β-turns)].
ΑΝΑΦΟΡΕΣ
1. Liljas, A., Rossman, M.G. (1974) Ann.Rev.Biochem., 43, 475-507.
2. Mathews, B. (1976) Ann. Rev. Phys. Chem., 27, 493-523.
3. Anfinsen, C.B., Haber, E., Sela, M . and White, F.H., Jr (1961) Proc. Nat. Acad. Sci. U.S.A., 47, 1309-1314.
4. Chou, P.Y., Fasman, G.D. (1974) Biochemistry, 13, 211-222.
5. Chou, P.Y., Fasman, G.D. (1974) Biochemistry, 13, 222-245.
6. Dufton, M.J., Hider, R.C. (1977) Biochemistry, 115, 177-193.
7. Chou, P.Y., Fasman, G.D. (1977) Biochemistry, 135-175.
8. Chou, P.Y., Fasman, G.D. (1977, June) Trends in Biochemical Sciences, p.128.
9. Levitt, M. and Greer, J. (1977) J.Mol.Biol., 114, 181-293.
10. B Rost: PHD: predicting one-dimensional protein structure by profile based neural networks. Methods in Enzymology, 266, 525-539, 1996.
11. B Rost, and C Sander: Prediction of protein secondary structure at better than 70% accuracy. J Molecular Biol, 232, 584-599, 1993.
12. B Rost, and C Sander: Combining evolutionary information and neural networks to predict protein secondary structure. Proteins, 19, 55-77, 1994.
13. Jones, D. T. (1999) Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 292: 195-202.
14. Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W. and Lipman., D.J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25: 3389-3402.
15. Cuff J. A. and Barton G. J. (1999) Evaluation and Improvement of Multiple Sequence Methods for Protein Secondary Structure Prediction, PROTEINS: Structure, Function and Genetics. 34:508-519.
16. Hamodrakas, S.J., 1988. A protein secondary structure prediction scheme for the IBM PC and compatibles. CABIOS 4, 473–477.